
Analytical Science Trends for Cell-based Drugs Biomanufacturing
During the last decade, the cell and gene therapy or Advanced Therapy Medicinal Products (ATMPs) industry has evolved rapidly and with significant progress in controlling living cell products manufacturing, from product identity and purity analysis to the release stage. In this article, we will review some of the key challenges for product characterization and monitoring, and how physicochemical methods, when applicable, can leverage cell and gene therapy manufacturing.
Introduction
Advances in cell biology and genetic engineering have led to the development of personalized, targeted cell therapies for treating diseases. Within this class, immune cell therapies are among the most advanced and have already demonstrated clear evidence of clinical benefits in cancer[1] and infectious diseases.
Since the approval of the very first biologic—recombinant insulin in the early 80s, the development of more intricate recombinant proteins and monoclonal antibody treatments has been witnessed, many of which have risen to become top-selling drugs. However, the development of living cell therapies, such as Chimeric Antigen Receptor (CAR) T cell therapy, presents even greater challenges due to the complexity, size, and dynamic nature of human cells.
Human cells, being large and composed of millions of protein molecules, present a complex structure. This structure includes a membrane, cytoskeleton, and organelles, each performing with specific functions. Beyond their size and diverse biochemical composition, cell therapy products – which focuses on transplanted whole cells – are heterogeneous and dynamic, continually interacting and adapting to their environment. Consequently, it is extremely difficult to fully characterize their properties and functions, which can change based on manufacturing, storage, and administration processes. Each characterization assay only provides insight into a single or limited number of attributes at any given time. The task of choosing the most suitable cellular attributes to monitor, combined with the inherent constraints of each assay method, presents a significant challenge that the industry will overcome to fully harness the potential of cell therapy.
The different types of known immune cell therapies namely: CAR-T cells, T cell receptor (TCR) T cells, tumor-infiltrating lymphocytes (TILs), natural killer (NK) cells, B cells, engineered macrophages, and dendritic cells are among the cell-based immunotherapies under development, also known as adoptive cell therapies (Figure 1).
Cell therapies can be autologous (using the patient’s own cells) or allogeneic (using cells from a donor), each with its advantages and disadvantages.
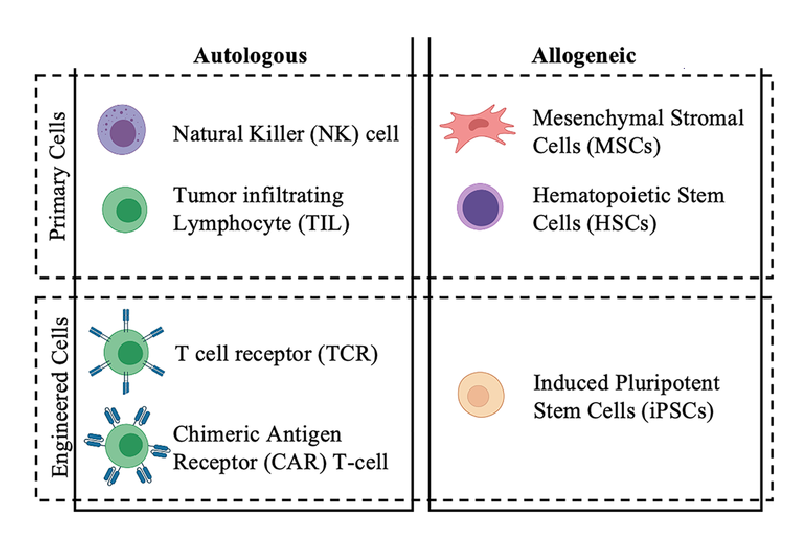
Figure 1: Different types of cell therapies derived from primary cells (i.e., cell procured from donor and not genetically modified) or engineered cells (i.e., cells procured from donor and subsequently genetically reprogrammed to specify or enhance the therapeutic application) [11]
Most of cell and gene therapies originate from an initial cell source obtained from donor tissue. Donor-to-donor variation poses a substantial obstacle in achieving a consistent, predictable manufacturing output. In the case of autologous therapies, additional variation arises due to patients presenting with varying degrees of illness severity or having undergone multiple treatments before providing tissue. These factors generate significant challenges in selecting appropriate attributes to test and overcoming assay method limitations to realize cell therapy’s full potential.
Currently, the US Food and Drug Administration (FDA) has approved five CAR T-cell products (namely, tisagenlecleucel, axicabtagène ciloleucel, brexucabtagene autoleucel, lisocabtagène maraleucel, idecabtagene vicleucel) for treating various blood cancers[2].
Challenges in characterizing cell therapies
During cell and gene therapy production (figure 2), major challenges emerge when ensuring the viability, proliferation (ie replication of cells for expansion), and quality of the cells throughout the manufacturing process[3].
Unlike biopharmaceutical, where proteins and antibodies are end products, cell therapies are expanded ex vivo, or cultured to generate the manufactured product. Because cells need to remain viable and highly responsive to their surrounding conditions, tailored culture media with supplements that provide nutrients for the survival and proliferation of the cells are requisite for the culture expansion process[4]. As a limitation, raw materials such as serum supplements derived from allogeneic sources further add variabilities to the manufacturing process, mostly due to the undefined components and lot-to-lot differences. Additionally, each cell therapy product is generated for a specific indication.
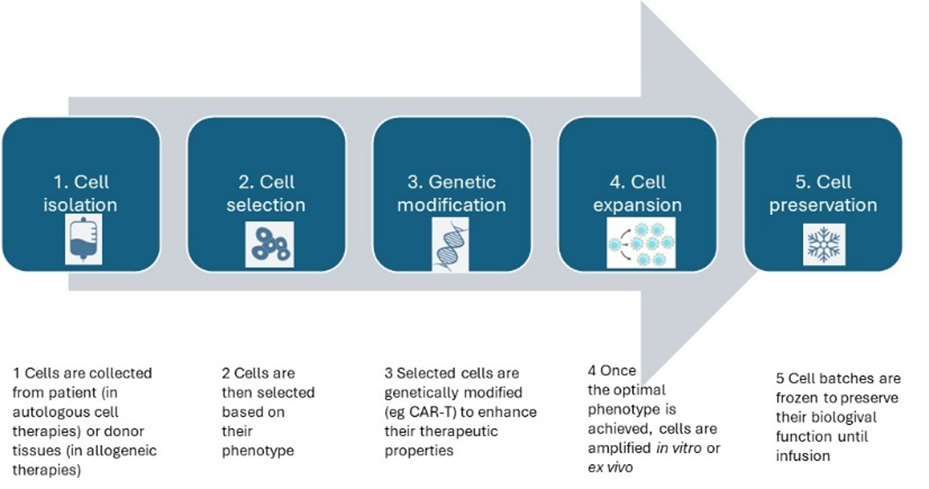
Figure 2: Immune cell therapy product development
Analytical Testing Strategies
Assay lifecycle development for traditional biopharmaceuticals such as vaccines and monoclonal antibodies (MAbs) has a clearly defined pathway, from preclinical method selection, development, and optimization through the milestones in preclinical phase trials, and finally to postlicensure method evaluations, comparability, and improvements (figure 3). The analytical development roadmap for ATMPs such as CAR-T cell therapies and gene therapies are more complex and can present many challenges along the way.
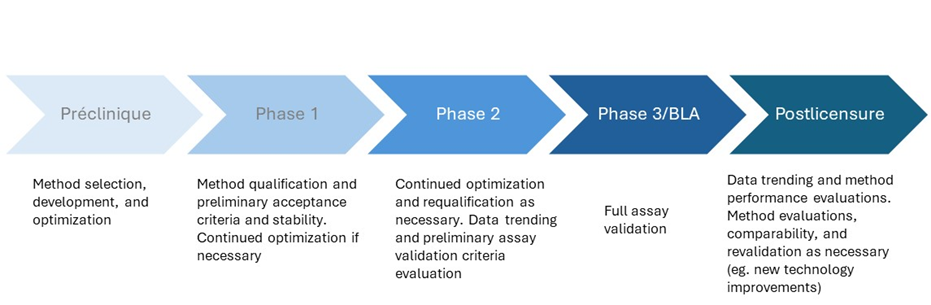
Figure 3: shows the typical analytical development lifecycle for well-established biopharmaceuticals.
With CAR-T cell and other autologous therapies, each patient is different, so each product is necessarily different. Clinical development needs to be in lockstep with chemistry, manufacturing, and controls (CMC) to expedite timelines efficiently. Analytical methods development is a continuous process and it is essential to understand what can be or not controlled.
Potential challenges include patient sample variability, small lot sizes, potency needs, and product stability concerns. Moreover, autologous cell therapies require rapid release, and analysts must adjust testing methods to meet short timelines. Some drug products will go to phase 1 after the preclinical stage, which allows for additional assay development time, but lifesaving drug addressing most medical needs, might go directly to pivotal trial and development, shortening the available timeframe for analysis, with no opportunity to progress through several clinical trials.
Cell therapy developers have started implementing Quality by Design (QbD) principles in their product development process to provide high-quality products[5]. QbD is a systematic approach that emphasizes understanding the product and process, process control, and risk mitigation. The QbD process starts by defining the end goal or product and formulating a hypothesis on its mechanism of action (MoA), which informs the target product profile (TPP). Developers must then establish critical quality attributes (CQAs) and critical process parameters (CPPs) to control to achieve the desired clinical outcome.
Process Analytical Technologies (PAT) are the backbone of QbD, monitoring product critical quality attributes (CQAs) and modulating critical process parameters (CPPs). Unlike CQAs that directly correspond to product quality, CPPs (e.g., pH, temperature, dissolved oxygen [DO]) and Critical Material Attributes (CMA; e.g., signatures of input cells, ratios of cell subsets) influence CQAs, thus indirectly affect product quality[6]. PAT is a key element for understanding and capturing biological features once validated CQAs are determined[7] and is currently applied to monitoring and control of CPPs where sensing capabilities are key objectives.
Defining product specifications
Based on regulatory guidance[8] , characterization of ATMPS includes, physicochemical properties, biological activity, immunochemical properties, purity, impurities, identity, viability, genetic stability (if applicable), sterility, and safety. By definition, a specification consists of the assay or test, the protocol for performing it, and the numerical limits, ranges, or other observations that define a product’s acceptance criteria. These acceptance criteria can be divided into several categories—Identity, Purity, Potency, Safety, and Physicochemical properties.
Several guidelines are available for characterization and analytical methods, but only offer overarching principles and recommendations to help developers (figure 4).
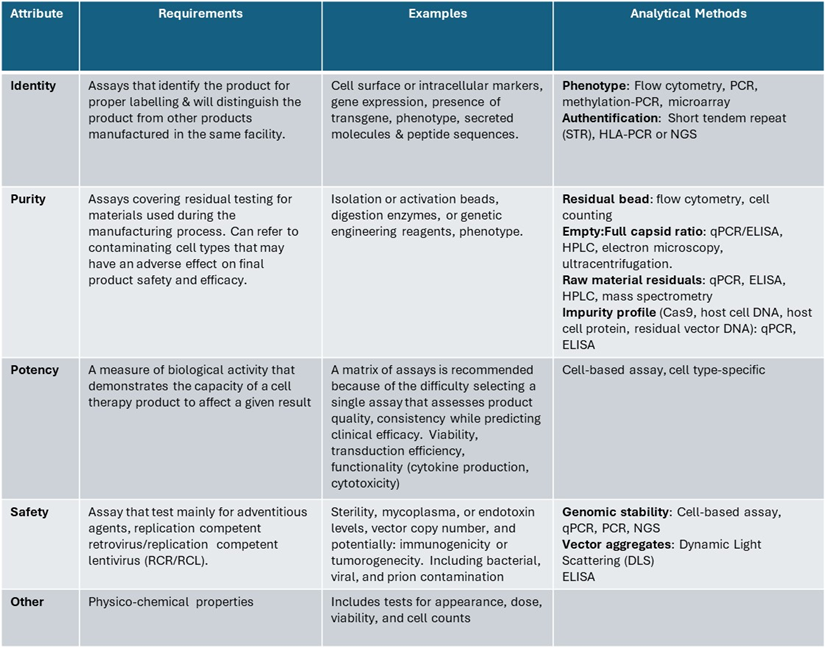
Figure 4: Overview of analytical methods used based on the required attributes
The primary analytical techniques used for cellular product characterization include cell counting, cell size, and viability analysis, using dyes and image-based analysis. Other CQAs, such as CAR expression, are assessed by phenotypic characterization using flow cytometry. Enzyme-linked immunosorbent assays (ELISA) are used to quantify the residual cytokines, with various cell activation agents used in the process. If the activation is bead-based, the residual bead amount in the final drug should be quantified – microscopy-based methods quantify the bead agents. Finally, PCR-based methods are used to quantify the vector copy number in gene-modified cell therapies.
Developing rapid, reliable, and validated analytical assays for microbiological agents is a key challenge for cell therapy.
Another key analytical challenge is establishing in vitro potency assays that represent the therapy’s mechanism of action in vivo, which is either undefined or far more complex than one would encounter with small molecules or monoclonal antibodies. Plus, cellular products cannot be terminally sterilized. The regulatory agencies understand this challenge and have issued specific guidance[9] to address the raw material (donor cells, master, and working cell banks) and in-process and release testing parameters.
In the final stage of product release, it is essential to conduct tests to ensure sterility (confirming the absence of microbiological and incidental agents), ensure identity (product characterization), assess purity (testing for acceptable limits of contaminants), and evaluate potency. Autologous therapy products are produced on a per-patient basis and are administered back to the patient as promptly as possible. Throughout the manufacturing process, aseptic methods are strictly maintained, and sterility tests may be performed 48 to 72 hours before the final cell harvest/formulation or after the last feeding of the cell culture.
A large panel of Physicochemical Methods in Cellular and Gene Therapies
Physicochemical methods play an important role in the characterization of viral vectors and ancillary materials. Vectors are used to transport the genetic material to the target cells (figure 5). The most commonly used viral vectors in gene and cell therapy are lentiviral vectors (LVV) and adeno-associated virus (AAV).
Multiple physicochemical techniques are used, and they are positioned to meet the unique needs of cell & gene therapy manufacturing process development, process and product characterization, and the quality control testing of these materials.
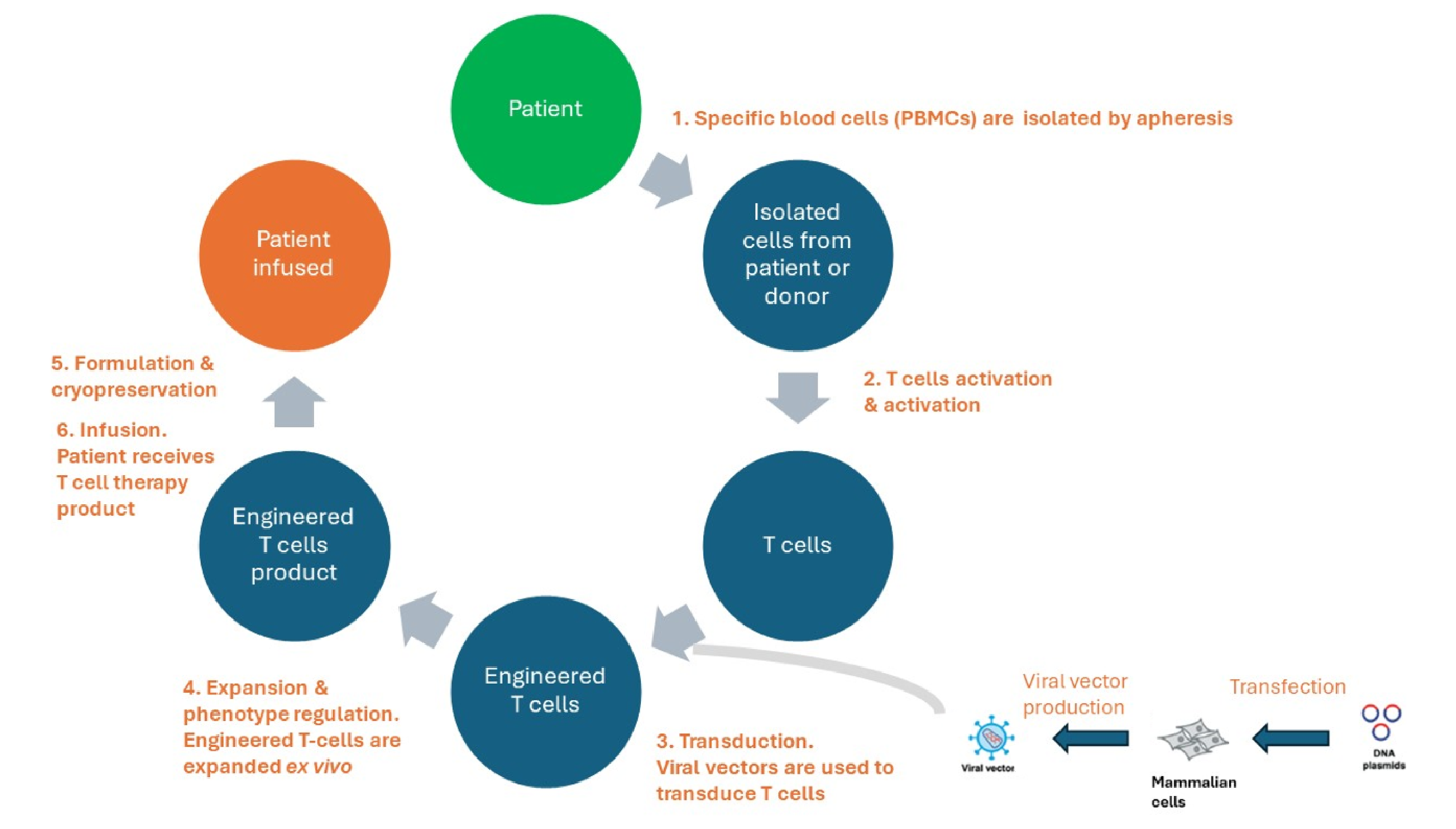
Figure 5: Lifecycle of T-cell therapy products
Comprehensive characterization of all ancillary materials employed in vector production, such as plasmids, cell culture media, or host cell line, is deemed essential (figure 5).
The rapid growth of cell & gene therapy calls for the adaptation of the diverse physicochemical methods to ensure the consistent quality of these critical materials. As analytical science advances to address increasing needs of product characterization, the progress of physicochemical methods will enable to further accelerate the drug development to ultimately serve patients in need.
Chromatography
Analytical chromatography is a well-established method for protein and small molecule analysis, Typically, chromatographic techniques can be used for adeno-associated virus (AAV) and other ancillary materials in the manufacturing process.
Ion exchange chromatography (IEC) can separate full AAV particles from empty ones, with improved detection limits, and within reduced run time methods, offering higher throughput compared with analytical ultracentrifugation (AUC).
Size-exclusion chromatography (SEC) has been developed as a sample preparation tool for LVV, removing protein impurities during sample preparation. When combined with tunable resistive pulse sensing technology, a single particle characterization method, SEC provides detailed profiling of the lentiviral vector, supporting vector process development.
Mass Spectrometry
Mass spectrometry (MS) is also a valuable tool for analyzing within high specificity. High-resolution MS techniques (HRMS) have improved the characterization of complex biotherapeutics, including gene and cell therapies. MS is particularly useful in the following applications:
- Host cell protein (HCP) characterization in vectors. It can particularly contribute to a better understanding on viral production and transduction efficiency.
- Post-translational modification of capsid proteins. MS approaches can help to identify deamidation sites and characterize their effects, especially in transduction efficiency.
Biophysical Methods
There are several biophysical methods for viral vector analysis, including analytical ultracentrifugation (AUC), multi-angle light scattering (MALS), and dynamic light scattering (DLS).
Imaging and Spectrometry Technologies
Imaging and spectroscopic technologies are also valuable tools for providing unique insights into the composition of products and raw materials, enabling the enumeration and visual identification of various particulates.
Monitoring with PAT techniques
In biopharmaceutical manufacturing, PAT is mostly used to monitor CPPs in upstream cell culture or microbial fermentation processes. More traditional sensor measurements in bioreactors include the measurement of pH, dissolved oxygen (DO), carbon dioxide, temperature, pressure, and capacitance. More advanced spectroscopic or chromatographic methods have been developed to perform in-line or at-line measurements of additional parameters such as metabolites or intermediates, host cell proteins (HCPs), nutrients, cell density, cell viability, aggregates, particulates, and product concentration or the simultaneous measurement of multiple analytes.
There is a strong trend for in-line monitoring (meaning the sample is analyzed directly in the process) as it allows a more dynamic, real-time analysis and fastest response times, especially in comparison to off-line measurements that would result in a significantly delayed response. A variety of in-line, non-destructive analytical technologies exist including infrared spectroscopy (IR), dynamic light scattering (DLS), and Raman spectroscopy[10]. Moreover, Fourier-transform infrared spectroscopy (FT-IR) could potentially enable in-line measurement of metabolic data, such as glucose and lactate values, which are currently measured using off-line or at-line enzyme-based sensors.
Implementing PAT[11] in cell therapy processing may also accelerate process development and characterization due to increased visibility and understanding of key process variables. These tools can provide insight into the influence of these variables on product attributes. Due to the limited quantity of patient sample available for autologous cell therapy processing and potential contamination risks associated with sampling events, on-line and in-line tools facilitate a closed, non-destructive means of process and product monitoring.
Concluding Remarks
Cell and Gene therapies present an entirely new paradigm in drug development. Furthermore, the cell therapy industry has evolved rapidly and with significant progress in controlling living cell products manufacturing. Facing important challenges, including the variability of human cells, the high cost of manufacturing, and the need for more sensitive and standardized assays, the field is moving from cell-based methods towards molecular-based methods. When applicable, molecular-based techniques offer greater sensitivity, standardization, and require less material for testing. Single-cell analysis is also becoming increasingly important, especially for gene-modified cell therapies, to understand transgene integration and ensure safety.
The ability to shorten vein-to-vein time and reduce production costs is particularly notable for autologous cell therapy, where the entire cost of manufacture is allocated to a single drug for a single patient.
Choosing a manufacturing organization that combine expertise in analytical science, chemistry, and cell manufacturing, such as CDMOs like SEQENS and CELLforCURE by SEQENS, can support immune therapies to the next level.
References
[1]R. L.-G. Haddock, N. Lumelsky, R. McFarland, K. Roy, K. Saha, J. Zhang, C. Zylberberg, NAM Perspect 2017, 1 ; I. Riviere, K. Roy, Mol. Ther. 2017, 25(5), 1067 ; C.C. Ma, Z.L. Wang, T. Xu, Z.Y. He, and Y.Q. Wei, Biotech.Adv. 40, 107502 (2020).
[2]A. Mullard, Nat. Rev. Drug Discov. 2017, 16(10), 669 ; A. Mullard, Nat. Rev. Drug Discov. 2017, 16(12), 818 ; FDA okays second CAR-T for Kite, Nat. Biotechnol. 2020, 38(9), 1012 ; D. S. Aschenbrenner, Am. J. Nurs. 2021, 121(6), 21 ; N. C. Munshi et al. N. Engl. J. Med. 2021, 384(8), 705.
[3]Y. Y. Lipsitz, N. E. Timmins, P. W. Zandstra, Nat. Biotechnol. 2016, 34(4), 393.
[4]S. Ghassemi, F. J. Martinez-Becerra, A. M. Master, S. A. Richman, D. Heo, J. Leferovich, Y. Tu, J. C. García-Cañaveras, A. Ayari, Y. Lu, A. Wang, J. D. Rabinowitz, M. C. Milone, C. H. June, R. S. O’Connor, Mol. Ther. Methods Clin. Dev. 2020, 18, 595.
[5]Juran JM (1992) Juran on quality by design: the new steps for planning quality into goods and services. Free Press
[6]Y. Y. Lipsitz, N. E. Timmins, P. W. Zandstra, Nat. Biotechnol. 2016, 34(4), 393.
[7]Guidance for Industry PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. (2004) FDA
[8]EMA. Guideline on human cell-based medicinal products. EMEA/CHMP/410869/2006. 2008. Available at: https://www.ema.europa.eu/en/human-cell-based-medicinal-products-scientific-guideline; EMA. Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. EMA/CAT/GTWP/671639/2008. 2012. Available at: https://www.ema.europa.eu/en/quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically-modified-cells-scientific-guideline; EMA. Guideline on the quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials. EMA/CAT/852602/2018. 2019. Available at: https://www.ema.europa.eu/en/quality-non-clinical-clinical-requirements-investigational-advanced-therapy-medicinal-products-clinical-trials
[9]FDA, Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs) (2008).
[10]Gerzon G, Sheng Y, Kirkitadze M. Process Analytical Technologies – Advances in bioprocess integration and future perspectives. Journal of Pharmaceutical and Biomedical Analysis. 2022;207:1143
[11]B. Wang, A. C. Bowles-Welch, C. Yeago, K. Roy, J. Adv. Manuf. Process. 2022, 4(1), e10106. https://doi.org/10.1002/amp2.10106




